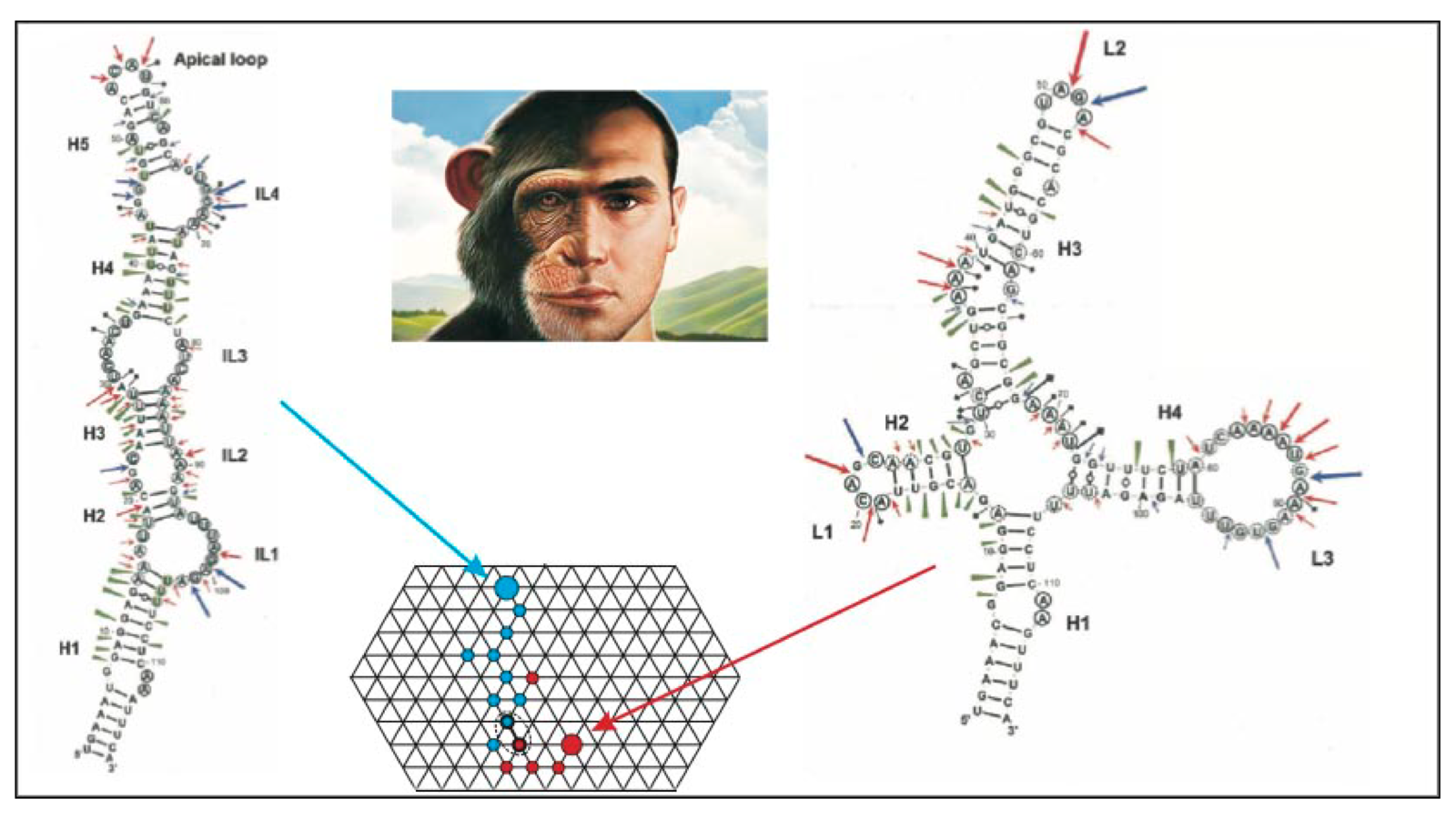
Genetic Data Information
DNA nucleotide information is transcribed into RNA, stabilized by molecular folding. In a plethora of interactions, it is the specific folding configuration and not the particular sequence of nucleotides that determines biological functionality. On one hand, genetic sequences like viruses as well as noncoding RNAs can mutate significantly, and still preserve their phenotype. On the other hand, sequences like riboswitches can switch between two phenotypes without any sequence change. Understanding the sequence-structure relation as well as their "hidden" information proves to be challenging.
Our research focuses on extracting information embedded in the sequence-structure pair that is not readily discoverable using sequence alignment. Specifically, we utilize the folding map, that takes RNA sequences to secondary structures, in order to understand sequence mutation impact on the phenotypic changes and vice versa. This line of work gives a unique insight into evolution dynamics involving both sequences and structures.
Research Highlights
- We developed Boltzmann samplers of sequence-structure pairs with various biologically meaningful constraints (Hamming distance filtration, genus filtration, etc.). These samplers facilitate the computational study of the shape-modulated evolution of biomolecules, as well as the design of functional noncoding genes.
- We studied the mutational robustness of let-7 miRNAs, in particular, with respect to multiple-point mutations. We showed that native let-7 genes exhibit higher mutational robustness compared to random inverse-folded sequences. We further demonstrated the connection between the robustness of let-7 miRNAs and cell differentiation across various organisms.
Ongoing Research
- We are investigating the mutational robustness of let-7 genes in cancer patients, comparing it to that of healthy people. Cancer cells are poorly differentiated compared to normal cells. Due to the important role of let-7 genes in cell differentiation, this line of work has potential application in predicting, preventing and treating cancers that corresponding to malfunctional let-7 genes.
- We are studying the energy-weighted density of the bi-compatible sequences of riboswitch alternative structures, revealing the phenotypic transition signals for the identification of riboswitches. The transitional signal also helps us to understand the irreversible sequence mutations in diseases.
Tools
-

This is a C program that samples RNA sequences from a given RNA secondary structure with a given Boltzmann distribution as well as with Hamming distance filtration. It takes a secondary structure, a reference sequence and a given distance as input, and generates RNA sequences that are Boltzmann distributed having the specified fixed Hamming distance to the reference sequence. Learn more.
-
 This is a C program that samples RNA sequences for two structures. It takes two RNA secondary structures having the same length as input, and generates sequences that are thermodynamically stable to the two input structures. Learn more.
This is a C program that samples RNA sequences for two structures. It takes two RNA secondary structures having the same length as input, and generates sequences that are thermodynamically stable to the two input structures. Learn more.